A new study led by researchers at the ICM has revealed that the environment likely influenced the evolution of bacterial predators in the ocean, which are essential for nutrient recycling.

A new study led by researchers from the Institut de Ciències del Mar (ICM) of Barcelona, with the participation of researchers from Norway, France and the United States, has revealed that the environment is key to the evolution of oceanic bacterial predators, single-celled eukaryotic microorganisms essential for the recycling of nutrients that will be used by higher trophic levels in marine food webs.
Specifically, the paper, published this week in the prestigious journal Proceedings of the National Academy of Sciences, finds that water temperature and the type of prey available to these predators strongly influence their evolution, and that this has contributed to the emergence of different species within the same family.
"The results suggest that these unicellular predators have evolved in a similar way to Darwin's finches in the Galapagos Islands, that is, from a common ancestor, adapting to different ocean temperatures or different diets", explains Ramiro Logares, ICM researcher and lead author of the study.
The results highlight the importance of understanding the ecology and evolution of these organisms at the species level, something that is not particularly easy considering that most of them cannot be cultured in the laboratory, which makes it very difficult to study them.
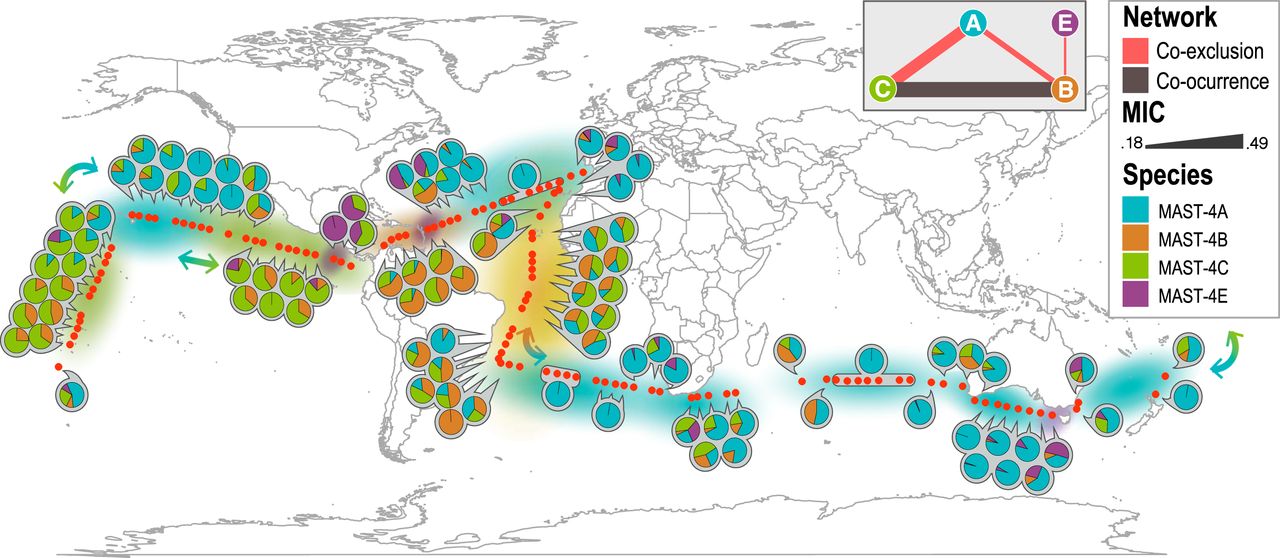
Among the microorganisms not cultured yet are most of the unicellular eukaryotes with predatory capacity -the ones studied in this paper- which are considered essential in the ocean because they establish predator-prey interactions at the base of the marine food web. These microorganisms are often categorised within the same functional group despite having different ecologies and evolutionary histories.
"Apart from the added difficulty of obtaining a quality genome, without a culture it is extremely difficult to test any kind of hypothesis that requires the study of an organism under different environmental conditions", states Francisco Latorre, also a researcher at the ICM and first author of the study.
To carry out the study, researchers studied four species belonging to the same family of uncultured organisms obtained from water samples taken during the Malaspina and Tara Oceans global expeditions, both held between 2009 and 2013. During these expeditions, thousands of samples of microorganisms were collected from the world's oceans.
Various techniques such as Single-Cell Genomics, metabarcoding, metagenomics and metatranscriptomics were used for their analysis. The researchers also used bioinformatics to determine the genomes of these species, reveal their metabolic potential and the distribution and expression of their genes in the global ocean.
The aim was to determine genomic differences between the species studied, as these could be key to understand the metabolic adaptations that these species have undergone and the existing relationships between these adaptations and their biogeographical distributions. Finally, researchers carried out a reconstruction of the genomes of these four species of bacterial predators.
"When we discovered the existence of this family of bacterial predators through environmental sequencing more than 20 years ago, we could not have imagined that we would come to know with this level of detail the genomes, biogeographies and evolutionary histories of the different species that form the family", adds Ramon Massana, another ICM researcher who participated in the work.
The results of this study may have implications for understanding the effects of global change on ocean microorganisms. According to the research team, an increase in ocean temperatures could change the geographical distributions of these predators, with potentially difficult-to-predict effects at the base of marine food webs.
In the future, the authors intend to further study the different populations of these predators in the global ocean and in a time series using metagenomics data in conjunction with nucleotide variant analysis. This will allow to understand possible adaptive changes at the gene level that may give rise to different populations.